Working with AtomGroups¶
A AtomGroup has a large number of methods attributes defined that provide information about the atoms such as names, indices, or the coordinates in the positions attribute:
>>> CA = u.selectAtoms("protein and name CA")
>>> r = CA.positions
>>> r.shape
(214, 3)
The resulting output is a numpy.ndarray. The main purpose of MDAnalysis is to get trajectory data into numpy arrays!
Important methods and attributes of AtomGroup¶
The coordinates positions attribute is probably the most important information that you can get from an AtomGroup.
Other quantities that can be easily calculated for a AtomGroup are
the center of mass centerOfMass() and the center of geoemtry (or centroid) centerOfGeometry() (equivalent to centroid());
the total mass totalMass();
the total charge totalCharge() (if partial charges are defined in the topology);
the radius of gyration
\[R_\mathrm{gyr} = \sqrt{\frac{1}{M}\sum_{i=1}^{N} m_i(\mathbf{r}_i - \mathbf{R})^2}\]with radiusOfGyration();
the principal axes \(\mathbf{p}_1, \mathbf{p}_2, \mathbf{p}_1\) from principalAxes() via a diagonalization of the tensor of inertia momentOfInertia(),
\[\Lambda = U^T I U, \quad \text{with}\ U=(\mathbf{p}_1, \mathbf{p}_2, \mathbf{p}_3)\]where \(U\) is a rotation matrix whose columns are the eigenvectors that form the principal axes, \(\Lambda\) is the diagonal matrix of eigenvalues (sorted from largest to smallest) known as the principal moments of inertia, and \(I = \sum_{i=1}^{N} m_i [(\mathbf{r}_i\cdot\mathbf{r}_i)\sum_{\alpha=1}^3 \mathbf{e}_\alpha \otimes \mathbf{e}_\alpha - \mathbf{r}_i \otimes \mathbf{r}_i]\) is the tensor of inertia.
Exercises 3¶
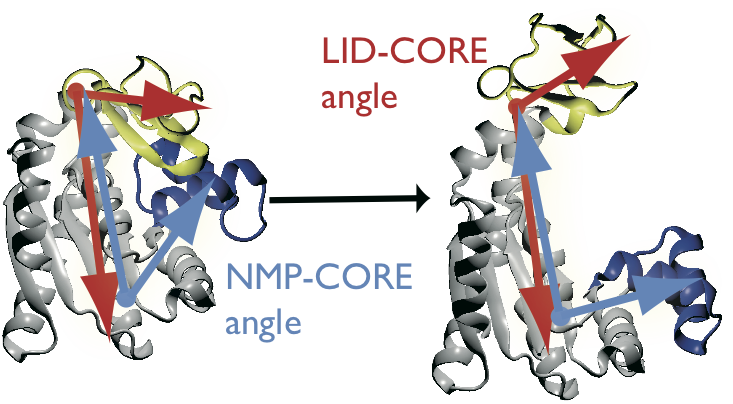
AdK consists of three domains:
- CORE residues 1-29, 60-121, 160-214 (gray)
- NMP residues 30-59 (blue)
- LID residues 122-159 (yellow)
Calculate the center of mass and the center of geometry for each of the three domains.
What are the distances between the centers of mass?
(Hint: you can use numpy.linalg.norm() or use a function like veclength() that you defined previously)
Does it matter to use center of mass vs center of geometry?
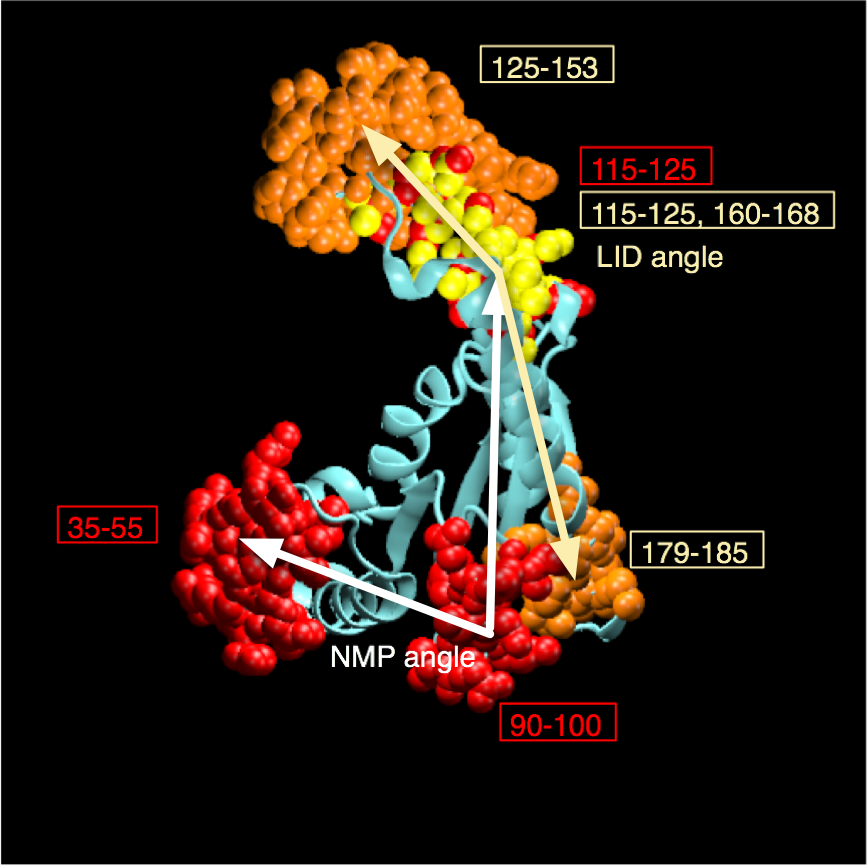
AdK undergoes a conformational transition during which CORE and LID move relative to each other. The movement can be characterized by two angles, \(\theta_\text{NMP}\) and \(\theta_\text{LID}\), which are defined between the centers of geometry of the backbone and \(\text{C}_\beta\) atoms between groups of residues [Beckstein2009]:
- definition of \(\theta_\text{NMP}\)
- A: 115-125, B: 90-100, C: 35-55
- definition of \(\theta_\text{LID}\)
- A: 179-185, B: 115-125, C: 125-153
The angle between vectors \(\vec{BA}\) and \(\vec{BC}\) is
Write a function theta_NMP() that takes a Universe as an argument and computes \(\theta_\text{NMP}\):
Use the following incomplete code as a starting point:
import numpy as np from np.linalg import norm def theta_NMP(u): """Calculate the NMP-CORE angle for E. coli AdK in degrees""" A = u.selectAtoms("resid 115:125 and (backbone or name CB)").centerOfGeometry() B = C = BA = A - B BC = theta = np.arccos( return np.rad2deg(theta)Write the function in a file adk.py and use ipython %run adk.py to load the function while working on it.
Test it on the AdK simulation (actually, the first frame):
>>> theta_NMP(u) 44.124821
Add the corresponding function theta_LID() to adk.py.
Test it:
>>> theta_LID(u) 107.00881
Processing AtomGroups¶
You can directly write a AtomGroup to a file with the write() method:
CORE = u.selectAtoms("resid 1:29 or resid 60:121 or resid 160:214")
CORE.write("AdK_CORE.pdb")
(The extension determines the file type.)
You can do fairly complicated things on the fly, such as writing the hydration shell around a protein to a file
u.selectAtoms("byres (name OW and around 4.0 protein)").write("hydration_shell.pdb")
for further analysis or visualization.
You can also write Gromacs index files (in case you don’t like make_ndx...) with the write_selection() method:
CORE.write_selection("CORE.ndx", name="CORE")